RNA-Sequencing이란?
RNA-Sequencing 기술은 NGS 기반으로 시퀀싱을 하는데, DNA에서 전사된 RNA를 시퀀싱하는 것을 말합니다. RNA 수준에서 알 수 있는건 Alternative splicing이나 유전자들의 발현량 등을 확인할 수 있죠.
이번 게시글에서는 대표적인 Bulk RNA-Seq 데이터를 분석하는 과정에 대해서 알아보겠습니다.
Single cell RNA-Sequencing 데이터 분석 과정은 나중에 알아보도록할게요!
NGS란?
서론 이번 게시글에서는 Next Generation Sequencing (NGS)가 무엇인지에 대해 알아보도록 하겠습니다. NGS는 한국어로 해석하면 차세대 염기서열 분석법이라고 부릅니다. 물론 현재는 차세대라고 하기에
develsw.tistory.com
RNA-Sequencing 분석 과정
NGS 기반의 시퀀싱을 하게 되면 결과가 FASTQ 파일로 나옴을 알고 계실겁니다.
[바이오인포매틱스] Fastq 파일형식
FASTQ format 'AGCT'로 표현되는 생물학적 sequence와 아스키 코드 (ASCII) 문자로 표시된 각 염기에 대한 quality (Phred)score를 모두 저장하기 위한 Text-based format으로 주로 NGS 기술로 생산된 sequenci..
develsw.tistory.com
그럼 이 Fastq 파일을 어떻게 분석해야지 우리가 원하는 목적인 유전자 발현량을 카운트할 수 있을까요? 그 과정을 한 번 이번 게시글에서 간략하게 알아보고, 각 과정들은 각각의 게시글로 다뤄보도록 하겠습니다.
Quality Control
우선, 데이터 분석을 하는 사람이라면 데이터를 받았을 때, 가장 먼저 데이터의 퀄리티를 확인해야됩니다. 질 낮은 데이터로 분석을 하게 된다면 그 결과는 신뢰할 수 없으니까요?
Fastq 파일에는 각 염기에 대한 퀄리티점수가 포함되어 있습니다. 퀄리티 점수 등을 기반으로 Fastq 파일의 퀄리티를 분석하는거죠. 만약 우리가 이걸 모르고 분석을 진행해서 나중에 데이터의 퀄리티가 좋지 않다는걸 알았을 때는 너무 많은 시간을 낭비하게 되는거죠.
다행히 저희가 직접 QC를 진행하지는 않고, QC 해주는 소프트웨어가 많이 개발되어 있습니다.
- FastQC
- RNA-SeQC
등등이 있습니다.
대표적으로 주로 사용하는게 FastQC입니다.
Babraham Bioinformatics - FastQC A Quality Control tool for High Throughput Sequence Data
www.bioinformatics.babraham.ac.uk
이 툴은 사용법도 간단합니다. 단순히 fastq 파일을 툴의 인풋으로 넣으면 알아서 QC를 해주고, 결과 레포트를 알려줍니다. 아래와 같이요.
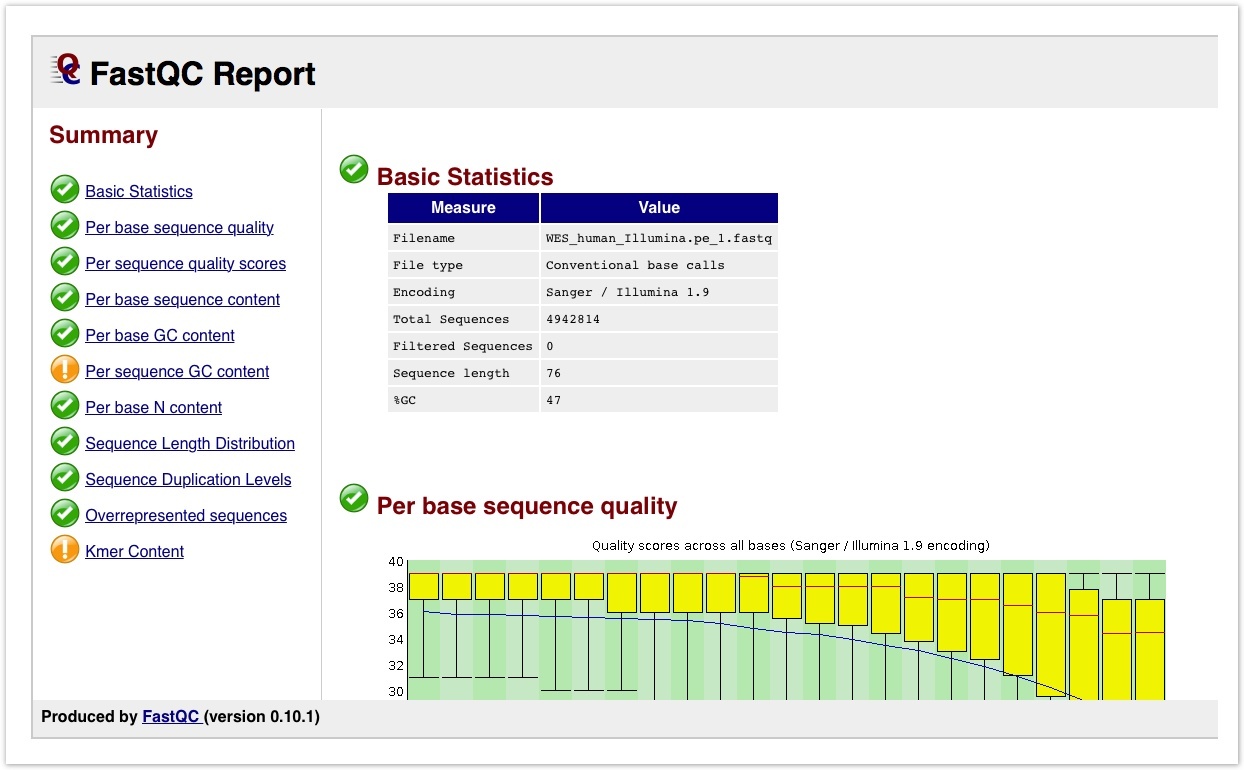
우리는 이 레포트를 기반으로 원본 데이터의 퀄리티를 판단할 수 있습니다.
Trimming adapters
NGS과정 중에 시퀀서에 Fragment들이 들어가게 되면 내부에서 어댑터 시퀀스를 제거하는 과정이 있습니다. 하지만, 이 과정에서 어댑터 시퀀스가 제대로 제거되지 않았을 경우가 있겠죠? 또한 우리가 이전 QC 과정에서 데이터의 품질에 대한 확인을 거쳤지만, 여전히 퀄리티가 좋지 않은 시퀀스들이 있습니다. 이런 질 낮은 염기들은 나중에 분석과정에서 아웃라이어에 영향을 주겠죠. 그렇기 때문이 이런 경우를 대비하기 위해 제거하는 소프트웨어가 필요하게 됩니다.
다음 과정은 어댑터 시퀀스를 자르는 과정과 퀄리티가 낮은 염기들을 제거하는 소프트웨어입니다.
대표적으로
Trimmomatic
USADELLAB.org - Trimmomatic: A flexible read trimming tool for Illumina NGS data
Trimmomatic: A flexible read trimming tool for Illumina NGS data Citations Bolger, A. M., Lohse, M., & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics, btu170. Downloading Trimmomatic starting on version 0.4
www.usadellab.org
Cutadapt
Cutadapt — Cutadapt 4.1 documentation
© Copyright 2010-2022, Marcel Martin Revision c1f3635b.
cutadapt.readthedocs.io
등이 있습니다.
Assembly
Assembly 과정은 NGS 과정에서 많은 조각들로 나눈 리드들을 다시 하나의 긴 시퀀스로 만들어주는 과정입니다. 이 과정에서 어셈블리는 두 경우로 나눌 수 있습니다. Reference 기반의 어셈블리와 De novo assembly 방법으로 나눌 수 있습니다.
Reference 기반의 어셈블리
Reference 기반의 어셈블리는 우리가 분석하고자 하는 종의 DNA reference genome이 존재한다면, 이 reference genome에 우리의 리드들을 붙여서 하나의 긴 시퀀스를 만들어가는 방법입니다. 대표적인 소프트웨어는 HISAT2, STAR 등이 있습니다. 자세한 사항은 따로 Assembly 관련 글에서 다뤄보도록 하겠습니다.
HISAT2
graph-based alignment of next generation sequencing reads to a population of genomes
DaehwanKimLab.github.io
GitHub - alexdobin/STAR: RNA-seq aligner
RNA-seq aligner. Contribute to alexdobin/STAR development by creating an account on GitHub.
github.com
De novo assembly
De novo assembly는 처음부터 조립한다는 의미를 가지고 있습니다. 즉 우리가 분석하고자 하는 종의 DNA reference genome이 존재하지 않는경우, 우리가 가지고 있는 데이터를 가지고 하나의 긴 시퀀스를 만드는 방법입니다. 대표적인 소프트웨어로는 Trinity 라는 소프트웨어가 있습니다. 이 역시 다음에 게시글로 다뤄보도록 하겠습니다.
GitHub - trinityrnaseq/trinityrnaseq: Trinity RNA-Seq de novo transcriptome assembly
Trinity RNA-Seq de novo transcriptome assembly. Contribute to trinityrnaseq/trinityrnaseq development by creating an account on GitHub.
github.com
Quantification
Quantification은 우리나라 말로는 정량화 한다는 의미입니다. 즉 위에서 우리가 어셈블리 하면서 특정 유전자의 부분에 많은 리드들이 붙어 있다면, 우리는 이렇게 생각할 수 있겠죠. 아 이 유전자가 많이 발현을 했구나? 이렇게 어떤 유전자가 얼마나 많은 발현을 했는지 알아보기 위해 정량화 단계를 거치게 됩니다.
정량화 해주는 대표적인 소프트웨어는 FeatureCounts 라는 소프트웨어가 있습니다.
The Subread package
Subread package: high-performance read alignment, quantification and mutation discovery The Subread package comprises a suite of software programs for processing next-gen sequencing read data including: Subread: a general-purpose read aligner which can ali
subread.sourceforge.net
오늘은 RNA-Sequencing 데이터의 분석 파이프라인에 대해서 알아봤습니다.
전체적인 flow는 아래와 같습니다.
QC ==> Trimming ==> Assembly ==> Quantification 의 과정입니다.
각 과정에 대해서는 좀 더 자세하게 다른 게시글에서 다뤄보도록 하겠습니다.
'공부 > 바이오인포매틱스' 카테고리의 다른 글
| PRS(Polygenic Risk Score)란? 계산 방법, 한계, R/Python 코드 예제 정리 (2) | 2025.08.31 |
|---|---|
| FASTQC 설치 방법 및 실행법 (0) | 2022.09.12 |
| Parallel-fastq-dump 설치하기 (0) | 2022.09.08 |
| NGS란? (0) | 2022.09.05 |
| 시퀀싱이란? (0) | 2021.10.11 |