서론
유전체 연구에서 질병 위험도를 평가할 때 가장 기본적인 도구 중 하나가 Polygenic Risk Score (다유전자 위험 점수, PRS) 입니다.
그런데, 여기서 먼저 짚고 넘어가야 할 개념이 있습니다. 바로 유전적 변이(variant) 입니다.
인간의 DNA는 약 30억 개의 염기서열(A, T, G, C)로 이루어져 있는데, 대부분은 사람마다 동일합니다. 하지만 약간씩 다른 부분이 존재하죠. 예를 들어, 어떤 사람은 특정 위치에서 A를 가지고 있는데, 다른 사람은 같은 위치에 G를 가지고 있을 수 있습니다. 이런 차이를 유전적 변이라고 부릅니다.
이러한 변이들이 쌓여서 결국 인간이나 생물의 다양한 모습(표현형) 을 만들어냅니다.
우리가 부모님과 비슷한 DNA를 공유하면서도 생김새나 성격이 다른 이유 역시, 이런 작은 변이들이 다르게 조합되었기 때문이죠.
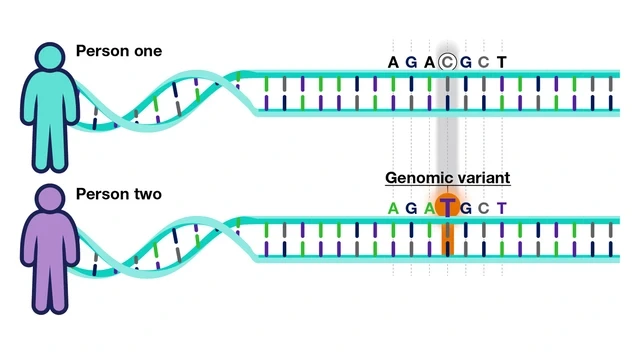
이 변이들 중 일부는 질병 발생 위험과 직접적으로 관련이 있습니다.
예를 들어, 특정 변이는 당뇨병 위험을 조금 더 높이고, 다른 변이는 심장질환 발생 확률에 영향을 줄 수 있습니다. 하지만 대부분의 복합질환은 한두 개의 변이로 설명되지 않고, 수백~수천 개의 작은 변이들이 조금씩 기여해서 나타납니다.
그래서 연구자들은 이런 변이들의 효과를 모두 합산해 개인별 위험 점수를 계산하는 방법인 PRS 를 개발했습니다.
쉽게 말해, “작은 주사위 눈금(변이 효과)이 여러 개 합쳐져서 최종 점수가 결정되는 방식” 이라고 생각하면 됩니다.
오늘날 PRS는 대규모 GWAS(Genome-Wide Association Study, 전장유전체연관분석)와 바이오뱅크 데이터 분석에서 기본적으로 사용되는 지표로 자리잡아, 질병 예측, 개인 맞춤 의학, 역학 연구 등 다양한 분야에서 활용되고 있습니다.
PRS의 정의 및 계산 방법
PRS를 이해하려면 먼저 SNP (Single Nucleotide Polymorphism, 단일염기다형성) 라는 개념을 알아야 합니다.
SNP는 가장 흔한 형태의 유전적 변이로, DNA 염기서열의 특정 위치에서 하나의 염기 (A, T, G, C)가 다른 염기로 바뀐 경우를 말합니다.
예를 들어, 대부분 사람에게서 특정 위치에 A가 있는 곳이 있는데, 일부 사람에게서는 G가 들어 있을 수 있습니다. 이런 차이를 SNP라고 부르며, 보통 사람 간 차이를 가장 잘 설명해주는 변이이기도 합니다.
SNP는 매우 흔해서, 평균적으로 약 1,000개 염기마다 1개 꼴로 존재합니다. 이 작은 차이들이 모여서 사람마다 질병 위험이나 약물 반응이 달라지게 되는 거죠..
PRS는 GWAS(Genome-Wide Association Study, 전장유전체연관분석) 에서 추정된 효과 크기(effect size, β̂) 를 바탕으로 계산됩니다. GWAS에서는 수많은 단일염기다형성(SNP, Single Nucleotide Polymorphism) 들을 대상으로, 환자군(case)과 대조군(control) 간의 차이를 통계적으로 검정합니다.
그 결과 각 SNP마다 효과 크기(β̂) 와 p-value(통계적 유의성) 이 추정되는데, 이 β̂ 값이 PRS 계산의 핵심 가중치가 됩니다.
개인의 PRS는 각 SNP에서 가진 위험 대립유전자 수(Xj) 에 이 β̂ 값을 곱해, 모든 SNP에 대해 합산하여 구합니다.
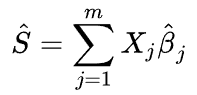
- PRS: 개인의 다유전자 위험 점수 (polygenic score)
- m: 고려하는 SNP의 개수
- X_j: 개인이 가진 j번째 SNP의 위험 대립유전자 개수 (0, 1, 2)
- β_j: GWAS에서 추정된 해당 SNP의 효과 크기 (effect size)
즉, “각 SNP의 위험 대립유전자 개수 × 그 SNP의 효과 크기” 를 모두 더한 값이 PRS가 되는 것입니다.
이 접근은 사실상 회귀 분석(regression analysis) 의 일반적인 형태와도 동일합니다. GWAS에서 추정한 회귀계수(β̂)를 이용해 새로운 개인의 예측 점수를 만드는 과정이라고 볼 수 있죠.
PRS의 주요 활용
- 질병 예측: 당뇨, 심혈관 질환, 정신질환 등의 발병 위험도를 개인별로 예측
- 개인 맞춤 의학: 생활습관 관리, 예방적 검진 계획 수립, 약물 반응 예측 등에 활용
- 역학 연구: 집단 내/집단 간 질병 위험도의 차이를 분석하는 도구
PRS의 한계
집단 간 이식성 문제 (Trans-ethnic portability)
PRS는 보통 유럽인을 기반으로 한 GWAS 결과에서 만들어지는 경우가 많습니다.
하지만 이 점수를 한국인이나 아시아인 집단에 그대로 적용하면 정확도가 크게 떨어질 수 있습니다.
- 예시: 유럽 GWAS에서 심장질환 위험을 설명하는 SNP 100개를 모아 만든 PRS가 있다고 해봅시다.
이 SNP들의 효과 크기(β) 는 유럽 집단의 대립유전자 빈도(frequency)와 연관성을 반영한 값입니다.
그런데 아시아인에서는 같은 SNP의 빈도가 다르고, LD(연관불균형) 구조도 달라서 실제 기여도가 다르게 나타납니다. - 실제 연구에서, 유럽인 기반 PRS는 아시아인 집단에 적용했을 때 예측력이 절반 이하로 떨어진 사례가 많이 보고되었습니다.
즉, 각 집단의 고유한 유전체 구조를 반영하지 않으면 PRS의 일반화에 큰 한계가 있습니다.
환경 요인 미반영
PRS는 오직 유전적 요인만 반영합니다.
하지만 실제 질병 위험은 환경 요인과 생활습관에 크게 좌우됩니다.
- 예시: 어떤 사람이 PRS 상으로는 당뇨병 위험이 높게 나왔더라도,
평소에 건강한 식습관과 꾸준한 운동을 유지하면 실제 발병 확률은 낮아질 수 있습니다. - 반대로 PRS 점수는 낮더라도 흡연, 음주, 스트레스, 수면 부족 같은 생활습관이 누적되면 심혈관 질환 위험이 높아질 수 있습니다.
즉, PRS는 유전적 잠재 위험을 보여줄 뿐, 실제 질병 발생을 100% 예측할 수는 없습니다.
표준화 부족
아직까지 PRS를 계산하는 방법론은 연구마다 차이가 큽니다.
- 어떤 연구는 수만 개의 SNP를 모두 합산하고,
- 다른 연구는 p-value 기준으로 유의한 SNP만 선택해서 사용하기도 합니다.
- 또 어떤 곳은 LD 클럼핑(clumping), LDpred, PRS-CS 같은 베이지안 모델을 적용해 보정하기도 합니다.
이처럼 계산 과정이 표준화되지 않았기 때문에, 같은 질환에 대해서도 연구자마다 다른 PRS 값을 얻을 수 있습니다.
- 예시: 같은 사람의 데이터라도, 연구 A에서는 “상위 10% 위험군”으로 분류되는데,
연구 B에서는 “평균 수준”으로 나올 수 있습니다. - 이런 차이 때문에 PRS를 임상 진료에 바로 적용하기 어려운 상황이 지속되고 있습니다.
코드 예제
library(dplyr) # 데이터 처리와 파이프라인 연산을 위해 dplyr 패키지 로드
# 1) GWAS 요약통계 (Summary statistics)
# 각 SNP에 대해 효과 알렐(A1)과 효과 크기(beta)를 담은 테이블
sumstats <- tibble::tibble(
snp = c("rs1","rs2","rs3","rs4"), # SNP ID
a1 = c("A","G","T","C"), # 효과 알렐 (effect allele)
beta = c(0.10, 0.05, -0.12, 0.08) # GWAS에서 추정된 효과 크기 (effect size)
)
# 2) 개인별 유전형 데이터
# 각 개인(sid)이 각 SNP에서 효과 알렐(A1)을 몇 개 갖고 있는지 (0, 1, 2) 기록
geno <- tibble::tibble(
snp = rep(c("rs1","rs2","rs3","rs4"), times=3), # SNP ID 반복
sid = rep(c("s1","s2","s3"), each=4), # 개인 ID (s1, s2, s3)
x = c(2,1,0,1, # s1의 각 SNP에서 A1 개수
0,1,2,2, # s2의 각 SNP에서 A1 개수
1,0,1,0) # s3의 각 SNP에서 A1 개수
)
# 3) PRS 계산
prs <- geno %>%
inner_join(sumstats, by="snp") %>% # SNP 기준으로 개인 유전형(geno)과 요약통계(sumstats) 결합
mutate(w = x * beta) %>% # 각 SNP별 기여도 = (A1 개수 × 효과 크기)
group_by(sid) %>% # 개인별로 묶어서
summarise(PRS = sum(w), .groups = "drop") # 모든 SNP의 기여도를 합산 → PRS 점수 계산
print(prs) # 개인별 PRS 결과 출력
각 snp에 대한 Beta값이 이펙트 사이즈가 되는 것이죠! 그리고 해당 염기에서의 Allele의 수가 가중되는겁니다!
즉 특정 변이 Allele가 많을 수록 PRS가 증가하겠죠?
PRS는 보통 Plnik를 이용해서 구하게됩니다. GWAS 분석 이후에 구하게 되는거죠!
plink2 --bfile target --score sumstats.txt 1 2 3 header --out prs_result결론
Polygenic Risk Score는 단일 유전자의 돌연변이가 아닌 복합적인 유전 변이의 총합을 통해 질병 위험을 예측하려는 강력한 도구입니다. 아직은 인종 간 적용성, 환경 요인 반영, 계산 방법의 표준화 등 여러 과제가 남아있지만, 의생명 분야에서의 궁극적인 목표인 개인 맞춤 의학(Precision Medicine) 으로 가는 길에서 핵심적인 역할을 할 것으로 기대가 됩니다.
'공부 > 바이오인포매틱스' 카테고리의 다른 글
| FASTQC 설치 방법 및 실행법 (0) | 2022.09.12 |
|---|---|
| RNA-Sequencing 분석 파이프라인을 알아보자 (0) | 2022.09.11 |
| Parallel-fastq-dump 설치하기 (0) | 2022.09.08 |
| NGS란? (0) | 2022.09.05 |
| 시퀀싱이란? (0) | 2021.10.11 |